Welcome to WordPress. This is your first post. Edit or delete it, then start writing!
Currently browsing: Uncategorized
The hardest part of starting up is starting out

Update zur DiGA-Verordnung
Wir blicken auf ein Jahr DiGA-Verzeichnis zurück. 22 digitale Gesundheitsanwendungen sind inzwischen im Verzeichnis zu finden. 17 davon haben es allerdings nur vorläufig geschafft. Das heißt, diese Apps müssen in klinischen Studien erst noch unter Beweis stellen, dass ihr Nutzen die Risiken für den App-Anwender übersteigt. Dafür haben sie 12 Monate Zeit mit der Möglichkeit um 3 Monate zu verlängern. Bislang hat noch keine DiGA den Prozess von der Erprobung in die dauerhafte Listung geschafft. Mehr als die Hälfte der Anträge sind bereits an den Aufnahmekriterien der Zertifizierung gescheitert oder haben ihren Antrag zurückgezogen.
Für die Hersteller ist die Zertifizierung enorm ressourcenintensiv und schwierig – besonders in einer kurzen Frist von 12 Monaten. Hinzu kommt, dass die Vermarktung komplizierter ist.
Weitere Details sind in dem folgenden Artikel zu finden: https://www.healthon.de/blogs/2021/10/06/ein-jahr-diga-verzeichnis-erfolg-oder-ernuechterung
Weitere Verordnung für Medizintechnikhersteller
Die neue Gesundheits-IT-Interoperabilitäts-Governance-Verordnung (oder IOP Governance-Verordnung, kurz GIGV) soll am 01.Oktober 2021 in Kraft treten.
Was bedeutet Intraoperabilität im Gesundheitswesen? Laut MDR -Verordnung versteht man unter Interoperabilität die Fähigkeit von zwei oder mehr Geräten, einschließlich Software, vom gleichen oder von unterschiedlichen Herstellern Informationen auszutauschen und die ausgetauschte Information für die korrekte Ausführung einer spezifizierten Funktion zu nutzen, ohne den Inhalt der Daten zu verändern oder/und untereinander zu kommunizieren oder/und wie spezifiziert zusammen zu arbeiten.
Die enorme Zahl an Standards soll durch diese Verordnung eingedämmt werden. Die dann zur Verfügung stehenden Interoperabilitätsstandards sollen auch tatsächlich genutzt werden. Eine eigens eingerichtete Koordinierungsstelle soll dann mit Expertengremien, IOP_Expertenkreisen und Arbeitsgruppen herauszufinden, welche Interoperabilitätsstandards erforderlich sind, Priorisierung der Bedarfe vornehmen, Anforderungen an neue Interoperabilitätsstandards zu spezifizieren, Empfehlungen zu Standards herausgeben und auf einer Wissensplattform öffentlich zugänglich machen.
Von dieser Verordnung betroffen sind die Hersteller medizinischer Informationssysteme, DiGA-Hersteller und bestimmte Medizinproduktehersteller die Datenschnittstellen ihrer Produkte konform mit den festgelegten IOP-Standards gestalten müssen. Betroffen sind davon somit etwa Hersteller von Klinikinformationssystemen (KIS), Patientendatenmanagementsystemen (PDMS), Praxisinformationssystemen (PIS) und Radiologieinformationssystemen (RIS).
Es bleibt abzuwarten welchen Einfluss diese neue Verordnung auf Hersteller von Medizinprodukten (z. B. DiGA) und medizinischen Informationssystemen hat, z. B. durch neue bürokratische und zusätzliche Aufwände für Entwicklung und Regulatorik.
Lesen Sie den ausführlichen Beitrag auf:
Drohender Versorgungsengpass im Bereich Labordiagnostik
Die neue In-vitro-Diagnostik-Verordnung der EU tritt am 26. Mai 2022 in Kraft. Doch die Branche läutet die Alarmglocken. Einer Studie zufolge droht in der Labordiagnostik eine große Angebotsknappheit.
„Die Ergebnisse sind alarmierend. Wenn die EU-Kommission nicht reagiert, bahnt sich eine Gefährdung der flächendeckenden Versorgung mit Labordiagnostik an“, sagt VDGH-Geschäftsführer Dr. Martin Walger.
Acht Monate vor Inkrafttreten der neuen EU-In-Vitro-Diagnostik-Verordnung (IVDR) ist die Situation ähnlich wie vor Inkrafttreten der Medizinprodukte-Verordnung (MDR). Auch hier besteht ein enormes Risiko für die zukünftige Verfügbarkeit – in diesem Fall die medizinische Labordiagnostik. Zu diesem Schluss kommt der Verband der Diagnostika Industrie (VDGH) auf Basis der Ergebnisse einer aktuellen europäischen Studie. Die European Union Medical Device Competent Authority (CAMD) ist ein Zusammenschluss zuständiger nationaler Behörden, die eine Studie in Auftrag gegeben haben, um die Auswirkungen des geltenden Rechtsrahmens auf die Zulassung von In-vitro-Diagnostika (IVD) ab Mai 2022 zu untersuchen. Koordiniert wurde die Forschung von Medtech Europe, dem europäischen Verband für Medizintechnik. Die wichtigsten Ergebnisse sind:
- Zukünftig wird sich die Zahl der Produkte, die von der benannten Stelle zertifiziert werden müssen, verzehnfachen.
- Nach dem neuen Gesetz wurden nur sechs Stellen benannt. Im aktuellen Rechtsrahmen sind 18 benannte Stellen im Bereich der In-vitro-Diagnostik tätig.
- 53 % der IVD-Unternehmen haben noch keine benannte Stelle gefunden, bei kleinen und mittleren Unternehmen (KMU) sind es sogar 64 %.
- Nur 12% der für IVD-Produkte erforderlichen Zertifikate wurden ausgestellt.
- Im besten Fall werden bis zum 26. Mai 2022 61 % aller IVDs zertifiziert sein. Im schlimmsten Fall werden dies nur 24% aller IVDs ausmachen.
Die CAMD-Forschung deckt repräsentativ 90 % des diagnostischen Marktes ab. An der Befragung nahmen insgesamt 115 Unternehmen teil, davon mehr als ein Viertel aus Deutschland. Mehr als 70 % der Teilnehmer sind kleine und mittelständische Unternehmen. „Gerade die hohe Beteiligung kleiner und mittelständischer Unternehmen beschreibt die Branche wahrheitsgetreuer als bisherige Umfragen“, so Volger.
Klinische Studien im Rahmen der MDR erfolgreich meistern
Ein Hauptaugenmerk der MDR liegt auf dem Sicherheitsaspekt von Medizinprodukten – klinische Studien sollen helfen, ein vertretbares Nutzen-Risiko-Verhältnis für Anwender bzw. Patient zu ermitteln.
Wie plane ich eine klinische Studie? Welche Vorgaben sind bei der Durchführung einzuhalten? Gibt es unterschiedliche Vorgehensweisen für am Markt zugelassene oder nicht-zugelassene Produkte? All diese Fragen beantworten wir bei unserem Seminar zum Thema klinische Prüfungen von Medizinprodukten mit dem Koordinierungszentrum Klinische Studien (KKS). Hier erfahren Sie alles, was sie bei der Antragsstellung, beim Qualitäts- und Risikomanagement bis hin zur Inspektion und der behördlichen Überwachung beachten müssen.
Wann? Oktober 2021
Wo? HIT Campus Magdeburg
Themenschwerpunkte:
• Definitionen und Klassifizierung gemäß MDR 2017-745
• GCP-konforme Durchführung gemäß DIN ISO 14155
• Antragstellung
• Qualitäts- und Risikomanagement
• Inspektionen
• behördlicher Überwachung
Bei Interesse schreiben sie uns bitte eine kurze E-Mail an work@innomed-sachsen-anhalt.de. Das genaue Datum stimmen wir dann mit allen Interessenten ab.
Ein Jahr DiGA-Verzeichnis – was hat sich getan?
Das Bundesamt für Arzneimittel und Medizinprodukte (BfArM) blickt nun auf ein Jahr DiGA-Verzeichnis zurück. In der Pressemitteilung spricht Prof. Broich (Präsident des BfArM) von einem wichtigen Meilenstein in der sicheren digitalen Versorgung von Patienten in Deutschland. Mit Hilfe des DiGA-Verzeichnisses werden neue Therapieoptionen dynamisch vorangebracht und die Potenziale der Digitalisierung genutzt. In diesem Verzeichnis aufgeführte digitale Gesundheitsanwendungen können von Ärzten und Psychotherapeuten verordnet und von Krankenkassen erstattet werden.
Wie viele Anwendungen sind aktuell im Verzeichnis?
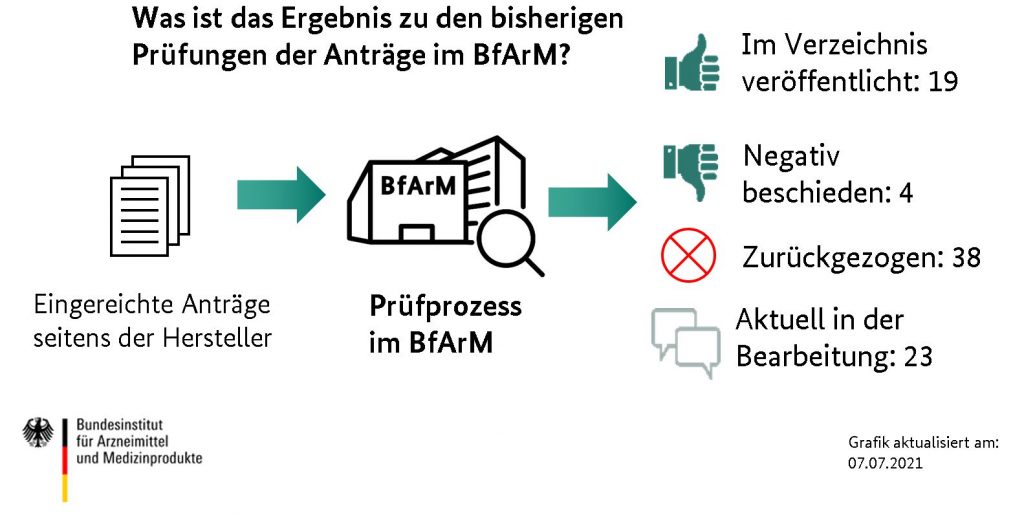
Unter Nutzung des Fast-Track-Verfahrens wurden inzwischen 84 Anträge auf Aufnahme ins DiGA-Verzeichnis gestellt, davon 60 zur vorläufigen und 24 zur dauerhaften Aufnahme. Bisher wurden 19 Anwendungen im Verzeichnis gelistet, 38 Anträge wurden von den jeweiligen Herstellern zurückgezogen und 4 vom BfArM negativ beschieden. Aktuell befinden sich 23 Anträge in der Prüfung.
Unser Vereinsmitglied HASOMED befindet sich mitten in den Vorbereitungen für den Antrag. Auch celloon mit der zur celloon-Gruppe gehörenden AID MEDWARE beschäftigt sich ausführlich mit dem Thema.
Welche Kriterien müssen erfüllt werden?
Natürlich sollte mit der Anwendung in erster Linie ein positiver Versorgungseffekt erzielt werden. Also eine Verbesserung des Gesundheitszustandes, eine Verkürzung der Krankheitsdauer, eine Verlängerung der Überlebensdauer oder eine Verbesserung der Lebensqualität. Krankenkassen möchten schließlich nur für Maßnahmen bezahlen, die den Patienten einen Mehrwert bieten. Zudem müssen definierte Anforderungen an die Sicherheit und Funktionstauglichkeit der Anwendungen erfüllt werden. Ebenso prüft das BfArM den Datenschutz, die Informationssicherheit, sowie die Qualität im Allgemeinen.





